Tutorials¶
Tutorial 1 – Polynomial Fitting¶
This example covers the basic structure and content of the
input file. The input file, e.g. skpar_optimise.yaml, would
typically reside in the invocation directory.
The models should have separate execution directories,
typically within the invocation folder.
The relevant files for the example are under skpar/test directory:
test_optimise.yaml, and the foldertest_optimise/, where the modeltest_optimise/model_poly3.pyis executed (and located)
The example can be run in the skpar/test directory by invoking:
skpar test_optimise.yaml,
assuming that skpar is installed.
Input YAML file¶
In this example we try to fit a 3-rd order polynomial to a few points extracted from such a polynomial.
The setup of SKPAR consists of 4 items:
- A list of objectives that steer the optimisation,
- A list of tasks necessary to evaluate the model,
- An optional dictionary of aliases (used in the task list) resolving to external executables,
- A configuration of the optimisation engine (parameters, algorithm, cost-function).
The corresponding yaml file, test_optimise.yaml reads:
The reference polynomial, the reference points from it (see ref: [...]
in the yaml file above, and the fitted 3-rd order polynomial may look as so:
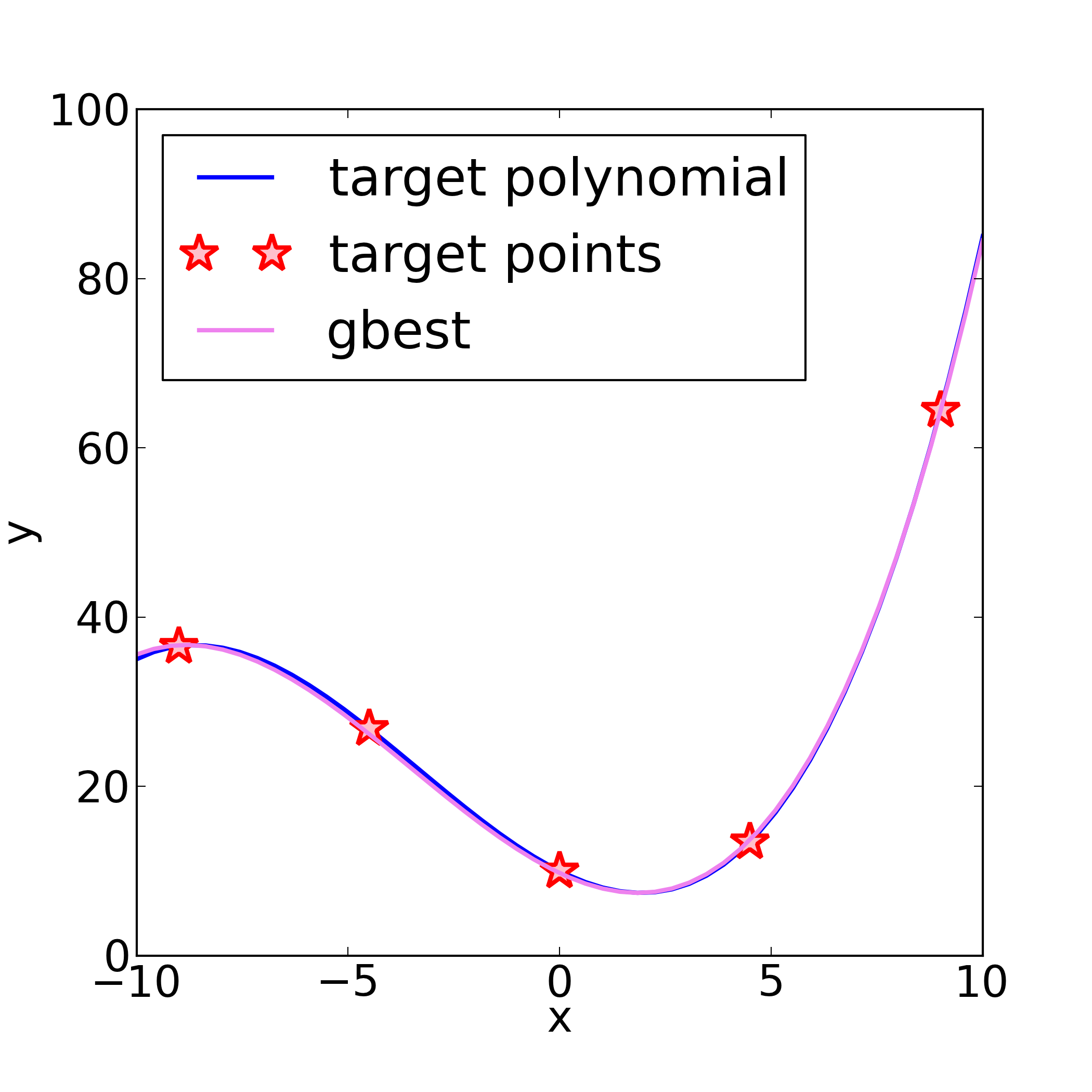
Comparison of reference and fitted (gbest) polynomials, and reference data points
What is happening?¶
Objectives:
A model named poly3 should yield data named yval, to be compared
against explicitly provided reference data ref: [...].
Fitness evaluation of this specific objective should be based on
root-mean-squared relative deviations, as stated after eval:.
Tasks (task-list):
At each iteration do:
- Set the environment by writing the parameters to
current.parand- substitute values in
template.parameters.pytoparameters.py, both files in./test_optimisefolder. (Note thatparameters.pyis not used by the model in this case.)
- Run the command
mypyin the./test_optimisefolder with- input file
model_poly3.py.
- Get the model data from
test_optimise/model_poly3_out.datand- associate it with the
yvalof modelpoly3in the model database.
Optimisation
Generate four parameters (with initial range as given by a pair of min/max values) according to particle swarm optimisation algorithm, using 4-particle swarm, evolving it for 5 generations.
Executables
Whenever a run-task requires mypy command, use python instead.
Tutorial 2 – Optimisation of electronic parameters in DFTB¶
Fitting to experimental data¶
A more elaborate example is fitting the electronic structure of bulk Si to match a set of experimentally known E-k points and effective masses.
Here we set three different objectives, each of them contributing several data items.
The corresponding skpar_in.yaml is below, with comment annotations:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 | executables: skgen: ./skf/skgen-opt.sh # script yielding an skf set bands: dftbutils bands # band-structure calculation # see documentation for dftbutils sub-package tasks: # Three types of tasks exist: # - set: [parmeter_file, working_directory, optional_template_file(s)] # - run: [command, working_directory] # - get: [what, from_sourse(dir, file or dict), to_destination(dict), optional_kwargs] # `what` is essentially a function name (see Get-Tasks dictionary) # ------------------------------------------------------------------------------ - set: [current.par, skf, skf/skdefs.template.py] # update ./skf/skdefs.py - run: [skgen, skf] # generate SKF-set - run: [bands, Si-diam] # run dftb+ and dp_bands in Si-diam - get: [get_dftbp_bs, Si-diam/bs, Si.bs, # get BS data and put in Si.bs model DB {latticeinfo: {type: 'FCC', param: 5.431}}] # must know the lattice for what follows - get: [get_dftbp_meff, Si.bs, # get electron effective masses {carriers: 'e', directions: ['Gamma-X'], # note: destination is ommitted, Erange: 0.005, usebandindex: True}] # hence update the sourse - get: [get_dftbp_meff, Si.bs, # get hole effective masses {carriers: 'h', directions: ['Gamma-X', 'Gamma-L', 'Gamma-K'], nb: 3, Erange: 0.005}] - get: [get_dftbp_Ek , Si.bs, # get eigen-values at special points {sympts: ['L', 'Gamma', 'X', 'K'], extract: {'cb': [0,1,2,3], 'vb': [0,1,2,3]}, align: 'Evb'}] objectives: - Egap: # item to be queried from model database doc: Band-gap of Si (diamond) # doc-string for report purposes (optional) models: Si.bs # model name must match destination of a get-tasks ref: 1.12 # explicit reference data in for this objective weight: 4.0 # relative importance of this objective # objective weight in the overall cost function eval: [rms, relerr] # objective function: RMS of relative error - effective_masses: # items to be queried here will be defined by doc: Effective masses, Si # explicit keys, since the reference data consists models: Si.bs # of key-value pairs ref: file: ./ref/meff-Si.dat # the reference data is loaded via numpy.loadtxt() loader_args: dtype: # NOTABENE: yaml cannot read in tuples, so we must # use the dictionary formulation of dtype names: ['keys', 'values'] formats: ['S15', 'float'] options: subweights: # individual data items have sub-weight within an objective dflt : 0.1 # changing the default (from 1.) to 0. allows us to consider me_GX_0: 1.0 # only select entries; alternatively, set select entries me_Xt_0: 0.0 # to zero effectively excludes them from consideration weight: 1.0 # objective weight in the overall cost function eval: [rms, abserr] # objective function: RMS of absolute error - special_Ek: doc: Eigenvalues at k-points of high symmetry models: Si.bs ref: file: ./ref/Ek-Si.dat loader_args: dtype: # NOTABENE: yaml cannot read in tuples, so we must # use the dictionary formulation of dtype names: ['keys', 'values'] formats: ['S15', 'float'] options: subweights: dflt : 0.1 # changing the default (from 1.) to 0. allows us to consider me_GX_0: 1.0 # only select entries; alternatively, set select entries mh_Xt_0: 0.0 # to zero effectively excludes them from consideration weight: 1.0 eval: [rms, relerr] optimisation: algo: PSO # algorithm: particle swarm optimisation options: npart: 2 # number of particles ngen : 2 # number of generations parameters: - Si_Ed : 0.1 0.3 # parameter names must match with placeholders in - Si_r_sp: 3.5 7.0 # template files given to set-tasks above - Si_r_d : 3.5 8.0 |
Fitting to DFT and experimental data¶
A yet another elaborate example is fitting the electronic structure of bulk Si using a combination of DFT-calculated band-structure and a set of experimentally known E-k points and effective masses.
This is mostly as before, but provision is made to fit against DFT calculations not only for equilibrium volume, but also for slightly strained primitive cell, e.g. within +/- 2% deviation from the equilibrium vollume.
Another important subtlety relates to the fact that the DFT-calculated band-gap is unphysically low (~0.6 eV for Si, rather than the experimentally known 1.12 eV), and the objectives aim to avoid this issue in the DFTB fit.
This is accomplished by creating a couple of separate objectives for fitting the shapes of the conduction and valence bands independently, along with the objective for reaching the experimental band-gap.
The corresponding skpar_in.yaml is below, with comment annotations:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 | config: templatedir: template workroot: ./_workdir keepworkdirs: true executables: skgen: ./template/skf/skgen-opt.sh bands: dftbutils bands tasks: - set: [skf/skdefs.template.py] - run: [skgen, skf] - run: [bands, Si-diam/100] - get: [get_dftbp_bs, Si-diam/100/bs, Si.diam.100, {latticeinfo: {type: 'FCC', param: 5.431}}] - get: [get_dftbp_Ek, Si.diam.100, {sympts: ['L', 'Gamma', 'X', 'K'], extract: {'cb': [0,1,2,3], 'vb': [0,1,2,3]}, align: 'Evb'}] objectives: - Egap: # if using : inside doc string, use '' or "" to surround the string doc: 'Si-diam-100: band-gap' models: Si.diam.100 ref: 1.12 weight: 5.0 eval: [rms, relerr] - bands: doc: 'Si-diam-100: valence band' models: Si.diam.100 ref: # This is bandstructure from VASP + vasputils, which makes it # in the same format as DFTB + dp_bands, i.e. bands are columns # in the file, with each row corresponding to a k-point, and # the k-points are indexed in column 1 (completely redundant) # The advantage of this is that the band with lowest energy # also has the lowest column index. # But for visualisation, bands span horisontally, and SKPAR # treats the bands-type of data as a 2D array where a band # is represented by a ROW in the array. # This is why, we must always transpose bands from dp_bands # or from vasputils, upon loading, and this is here accomplished # by the loader_args: {unpack: True} -- cf. numpy.loadtxt() for details. file: ~/Dropbox/projects/skf-dftb/Erep fitting/from Alfred/crystal/DFT/di-Si.Markov/PS.100/band/band.dat loader_args: {unpack: True} process: # indexes and ranges below refer to file, not array, # i.e. independent of 'unpack' loader argument rm_columns: 1 # filter k-point enumeration # rm_rows: [[41,60]] # filter K-L segment; must do the same with dftb data... but in dftb_in.hsd... # scale : 1 # for unit conversion, e.g. Hartree to eV, if needed options: # Indexes below refer to the resulting 2D array after loading, # transposing, and application of the rm_rows/rm_columns above. use_ref: [[1, 4]] # Fortran-style index-bounds of bands to use use_model: [[1, 4]] align_ref: [4, max] # Fortran-style index of band-index and k-point-index, align_model: [4, max] # or a function (e.g. min, max) instead of k-point subweights: # NOTABENE: # -------------------------------------------------- # Energy values are with respect to the ALIGNEMENT above. # If we want to have the reference band index as zero, # we would have to do tricks with the range specification # behind the curtain, to allow both positive and negative # band indexes, e.g. [-3, 0], inclusive of either boundary. # Currently this is *not done*, so only standard Fortran # range spec is supported. Therefore, band 1 is always # the lowest lying, and e.g. band 4 is the third above it. # -------------------------------------------------- dflt: 1 values: # [[range], subweight] for E-k points in the given range of energy # notabene: the range below is with respect to the alignment value - [[-0.1, 0.], 5.0] bands: # [[range], subweight] of bands indexes; fortran-style - [[2, 3], 1.5] # two valence bands below the top VB - [4 , 2.5] # emphasize the reference band # not supported yet ipoint: weight: 2.5 eval: [rms, relerr] - bands: doc: 'Si-diam-100: conduction band' models: Si.diam.100 ref: file: ~/Dropbox/projects/skf-dftb/Erep fitting/from Alfred/crystal/DFT/di-Si.Markov/PS.100/band/band.dat loader_args: {unpack: True} process: rm_columns: 1 # filter k-point enumeration # rm_rows: [[41,60]] # filter K-L segment options: use_ref: [5, 6] # fortran-style index enumeration: NOTABENE: not a range here! use_model: [5, 6] # using [[5,6]] would be a range with the same effect align_ref: [1, 9] # fortran-style index of band and k-point, (happens to be the minimum here) align_model: [1, min] # or a function (e.g. min, max) instead of k-point subweights: values: # [[range], subweight] for E-k points in the given range of energy - [[0.0, 2.5], 1.5] # conduction band from fundamental minimum to the band at Gamma - [[0.0, 0.1], 4.0] # bottom of CB and 100meV above, for good meff bands: # [[range], subweight] of bands indexes; fortran-style - [1, 2.5] # the LUMO only increased in weight; note the indexing # reflects the 'use_' clauses above weight: 1.0 eval: [rms, relerr] - special_Ek: doc: Si-diam-100, eigenvalues at special k-points models: Si.diam.100 ref: file: ./ref/Ek-Si.dat loader_args: dtype: # NOTABENE: yaml cannot read in tuples, so we must # use the dictionary formulation of dtype names: ['keys', 'values'] formats: ['S15', 'float'] options: subweights: dflt : 0.1 # changing the default (from 1.) to 0. allows us to consider Ec_G_0 : 0.5 # only select entries; alternatively, set select entries Ec_L_0 : 0.5 # to zero effectively excludes them from consideration Ec_X_0 : 2.0 Ev_L_0 : 2.0 Ev_K_0 : 2.0 Ev_X_0 : 2.0 weight: 1.0 eval: [rms, relerr] optimisation: algo: PSO # particle swarm optimisation options: npart: 2 # number of particles ngen : 2 # number of generations parameters: - Si_Ed : 0.1 0.3 - Si_r_sp: 3.5 7.0 - Si_r_d : 3.5 8.0 |